
Dra. Márcia de Oliveira Nicolini Nosralla, médica formada pela Universidade de S?o Paulo (USP), Mestre em Ciências da Saúde.
Têm Título de Especialista em Neurologia Infantil, pelo Hospital das Clínicas da Faculdade de Medicina da Universidade de S?o Paulo e de Neurofisiologia Clínica, com área de Habilita??o em Eletroencefalografia, pela Sociedade Brasileira de Neurofisiologia Clínica.
Fez estágio em Eletroencefalografia Neonatal, no Laboratoire d’électrephysiologie
du développement de l’H?spital de Port-Royal à Paris.
é membro titular da Sociedade Brasileira de Neurofisiologia Clínica, membro titular da Sociedade Brasileira de Neurologia Infantil e membro da Associa??o Internacional de Neurologia Infantil.
Há mais de 25 anos, dedica-se aos pacientes neurológicos e ao conhecimento científico.
A Neuropediatria ou Neurologia Infantil é a especialidade médica que estuda o processo de amadurecimento e do desenvolvimento do Sistema Nervoso de forma dinamica e contínua do recém-nascido até a adolescência.
As doen?as neurológicas na infancia se manifestam com particularidades próprias, pois interferem nesse processo.
Tem como fun??o o diagnóstico, prognóstico e tratamento das doen?as que envolvem o Sistema Nervoso (Central, Periférico e Aut?nomo) para reduzir as complica??es na idade adulta.
A mortalidade e a morbidade dos recém-nascidos de alto risco, isto é, aqueles que apresentam complica??es neonatais graves, como anoxia neonatal, crises convulsivas, desconforto respiratório, septicemia, e meningite, têm diminuído devido aos avan?os da obstetrícia e dos cuidados intensivos neonatais, mas as sequelas neurológicas, ainda constituem um sério problema para a saúde pública. Com isso, existe grande expectativa tanto dos familiares, quanto dos profissionais da saúde na determina??o do prognóstico dessas crian?as em rela??o à les?o cerebral. Por essa raz?o, há algum tempo, vários pesquisadores têm sido estimulados a determinar critérios para estabelecer o prognóstico neurológico nesses recém-nascidos (1-11).
O interesse por esse assunto aumentou quando, em 1932, Berger fez os primeiros estudos eletrencefalográficos em crian?as, onde observou as altera??es no padr?o da atividade elétrica cerebral de acordo com a faixa etária. Em 1938, novos conceitos foram formados com a Eletrencefalografia Neonatal (EEGN) (12)
A Eletrencefalografia Neonatal tem-se tornado uma técnica diagnóstica de fundamental importancia nesta faixa etária, por ser um método n?o invasivo e de baixo custo, que permite investigar a atividade elétrica cerebral nesta idade, com o aumento do nosso conhecimento na organiza??o e no funcionamento do cérebro, onde ocorre significativa matura??o do Sistema Nervoso Central (crescimento de dendritos e ax?nios, forma??o de sinapses, mieliniza??o e desenvolvimento bioquímico de neur?nios e glia). Além disso, é usada como complemento diagnóstico de doen?as neurológicas e para predi??o do prognóstico.
A Poligrafia Neonatal aumenta a acuidade da eletrencefalografia, pois também, avalia aspectos comportamentais, pois s?o associados os eletrodos n?o cerebrais ao E.E.G. (eletro-oculograma, eletromiograma, eletrocardiograma, fluxo aéreo nasal e cinta tóraco-abdominal para controle respiratório), para o estudo mais adequado do ciclo vigília-sono, que nesta faixa etária n?o é t?o claramente diferenciado como em adultos e crian?as maiores.
é utilizada, também para fazer o diagnóstico diferencial de patologias n?o cerebrais, como apnéia episódica de origem cardíaca. Por vezes, há a necessidade de realizar a monitoriza??o com o Vídeo-EEG para o estudo mais adequado de crises epilépticas.
Ao se laudar a Poligrafia Neonatal, determinando-a como normal, ou anormal, requer o conhecimento dos padr?es normais de cada idade gestacional, desde a prematuridade até os primeiros meses de vida.
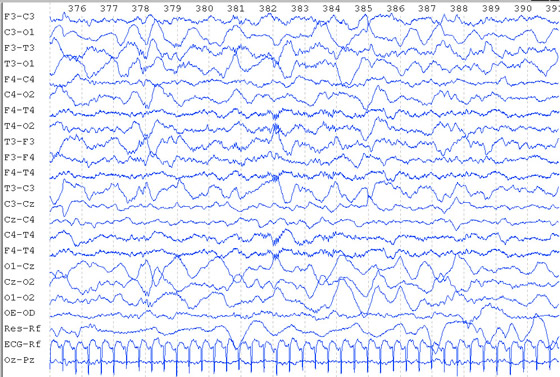 Delta-brush s?o ondas lentas na frequencia de 0,8 a 1,5 ciclos/segundo de 25 a 300
microvoltz, com superposi??o de ondas rápidas de 8 a 20 ciclos/segundos, com uma
amplitude de 10 a 100 microvoltz; seu pico de frequencia ocorre entre 32 e 34 semanas de
idade concepcional; s?o mais abundantes no sono-REM de crian?as entre 33-34 semanas de
idade concepcional e no sono n?o - REM em crian?as maiores; diminuem com o aumento das
idades gestacional e corrigida. Oz-Pz - eletromiograma.
Delta-brush s?o ondas lentas na frequencia de 0,8 a 1,5 ciclos/segundo de 25 a 300
microvoltz, com superposi??o de ondas rápidas de 8 a 20 ciclos/segundos, com uma
amplitude de 10 a 100 microvoltz; seu pico de frequencia ocorre entre 32 e 34 semanas de
idade concepcional; s?o mais abundantes no sono-REM de crian?as entre 33-34 semanas de
idade concepcional e no sono n?o - REM em crian?as maiores; diminuem com o aumento das
idades gestacional e corrigida. Oz-Pz - eletromiograma.
A Poligrafia Neonatal é de fundamental importancia para a complementa??o na avalia??o neurológica do recém-nascido, tanto de termo, como prematuro.
Devido a necessidade de se fazer um estudo mais adequado e contínuo dos bebês menores de 6 meses de idade, oferecemos o nosso servi?o com atendimento em Poligrafia Neonatal em nossa clínica.
1. Banker, BG e Larroche, J. Periventricular leukomalacia of infancy, a form of neonatal anoxic encephalopathy. Arch Neurol. 1962, 7: 386-410.
2. Anderson, C. e col. The normal E.E.G. of human newborn. Clin Neurophysiol. 1985, 2: 89-103.
3. Biagioni E. e col. Background E.E.G. activity in preterm infants: correlation of outcome with selected maturational feature. Electroencephalography and Clinical Neurophysiology. 1994, 91 : 154-61.
4. Blume, WT e Dreyfus-Brisac, C. Positive rolandic sharp wave in neonatal EEG: type and significance. Electroencephalography and Clinical Neurophysiology. 1982, 53: 277-82.
5. Cukier, F, André, M, Monod, N, Dreyfus-Brisac, C. Apport de le EEG au diagnostic des hémorragies intra-ventriculaires du prématuré. Rev EEG Neurophysioll Clin., 1972. 2: 318-22.
6. Lamblin MD e col. Indications of electroencephalogram in the newborn. Arch Pediatr. 2004 Jul; 11 (7): 829-33.
7. Lombroso, CT Neonatal EEG Polygraphy in normal and abnormal newborns. In: E. Niedermeyer and F. Lopes da Silva. Electroencephalography. Third Edition. Baltimore: Urban & Schwarzenberg: 1993: 803-75.
8. Murat, I. Intérêt discriminatif des pointes positives rolandiques. Contribution au diagnostic des hémorragies intraventriculaires. Thesis for the Doctor of Medicine, Academy of Paris, University René Descartes, Faculty of Medicine Cochin Port Royal, Paris, 1978.
9. Scher, MS. Midline electrographic abnormalities and cerebral lesions in the newborn brain. J Clin Neurol. 1988, 3: 135-46.
10. Tharp BR. Neonatal and pediatric electroencephalography. In: M.J. Aminoff (ed.), Electrodiagnosis in Clinical Neurology. Churchill-Livingstone, Edinburgh, 1980: 67-117.
11.Tharp BR, Cukier F e Monod N. The prognostic value of the Electroencephalogram in premature infants. Electroencephalography and Clinical Neurophysiology. 1981, 51: 219- 36.
12. Smith JR. The electroencephalogram during normal infancy and childhood. In Rhythmic activities present in the neonate and their subsequent development. J. Genet. Psychol. 1938, 53: 431-453.
Márcia de Oliveira Nicolini Nosralla
Neurofigiologista e Neurologia infantil
CRM: 51670
A forma??o do abscesso cerebral como complica??o única e exclusiva, decorrente de meningite bacteriana aguda ,diagnosticada e tratada corretamente e em pacientes sem fatores predisponentes, é uma eventualidade remota, isto é, o abscesso cerebral é raro como uma complica??o de meningite bacteriana, exceto em neonatos.
Ocorre em crian?as com:
1- doen?a cardíaca cianótica congênita;
2- após um procedimento neuro-cirúrgico;
3- traumatismos cranianos penetrantes;
4- focos infecciosos de localiza??o craniana (sinusite, otite, mastoidite, les?es supurativas da pele, foco dentário);
5- imunodepress?o;
6- doen?a pulmonar cr?nica;
7- endocardite bacteriana;
8- mal-forma??o (cisto dermal congênito com cisto dermóide).
Tanto a contamina??o hematogênica, como a difus?o direta ocorrem no abscesso cerebral.
Os abscessos s?o, geralmente nos hemisférios, mas podem ser achados no tronco cerebral ou no cerebelo.
A les?o, inicialmente é uma cerebrite, que pode persistir por várias semanas.
O edema que envolve a cerebrite, aumenta o efeito de massa. A cápsula do tecido de granula??o do processo inflamatório desenvolve ao redor da área infectada , após a fase da cerebrite.
O diagnóstico do abcesso cerebral é sugerido pelo curso sub-agudo: febre, cefaléia, confus?o mental, rebaixamento do nível de consciência, crises convulsivas ( focais ou generalizadas ), rigidez de nuca, papiledema, hemiparesia e/ou disfasia.
O efeito de massa do abscesso, pode obstruir o fluxo liquórico e desenvolver uma hidrocefalia.
Papiledema pode ocorrer, quando as suturas cranianas já estiverem fechadas. . Múltiplos abscessos podem ocorrer, principalmente, em doen?a cardíca cianótica congênitas.
O diagnóstico diferencial inclue: neoplasia, hematoma subdural e encefalite focal (herpes simples).
A Ressonancia Nuclear Magnética de cranio é o exame apropriado durante a cerebrite.
A Tomografia Computadorizada de Cranio e a Ressonancia Nuclear Magnética de Cranio s?o os exames complementares de escolha no abscesso maduro, pois revelam a característica capsular.
O E.E.G. pode ser normal, ou revelar anormalidades focais na regi?o do abscesso, com alentecimento, ou pontas. Pode, também mostrar descargas paroxísticas periódicas lateralizadas, ou pode ser difusamente lenta.
A pun??o do líquor deve ser evitada, devido ao risco de hernia??o secundária à hiperten??o intra-craniana. Quando o diagnóstico é dificil de ser feito e a neuro-imagem n?o sugere aumento da press?o intracraniana, a pun??o lombar pode ser considerada.
O liquor apresenta uma press?o aumentada. Pode mostrar-se normal, ou ter um leve aumento da celularidade, com predomínio de linfócitos, ou polimorfonucleares, eleva??o do nível de proteina e glicose normal. O Gram, geralmente é negativo e os organismos s?o identificados na cultura.
Bactérias anaeróbicas s?o achados em 70% dos abscessos cerebrais.
Streptococcus species, Staphylococcus species, e Bacteroides fragilis s?o as bactérias mais comuns. Em R.N., é o Proteus.
A conduta na hipertens?o intracraniana é a hiperventila??o, agentes osmóticos (manitol ), esteróides ( dexametazona ), e drenagem cirúrgica.
Antes da identifica??o do organismo, antibióticos apropriados para o provável agente etiológico, seriam administrados ( penicilina, cefotaxime e metronidazol).
O manuseio tradicional é cirúrgico (aspira??o ou completa extirpa??o), associado à antibióticoterapia a longo prazo.
A indica??o da pun??o do abscesso é discutível quando, está no estágio de cerebrite, quando há múltiplos abscessos, ou quando está numa área crítica.
A C.T. de Cranio ou a R.N.M. de cranio seriadas podem ser usadas para determinar a evolu??o na terapia medicamentosa. Sugere-se que a tomografia seja repetida a cada semana durante o tratamento. A seguir, a cada 2 a 4 semanas até a resolu??o da les?o, e após, a cada 2 a 4 meses até 1 ano. A diminui??o do tamanho da les?o come?ará entre 1 e 4 semanas.
N?o é necessário manter a terapia antimicrobiana até a normaliza??o tomográfica.
A terapia antimicrobiana é mantida por um período de 6 semanas.
Devido a dificuldade no diagnóstico e no manuseio do abscesso cerebral, a mortalidade e as seqüelas s?o freqüentes.
O prognóstico é pobre , quando múltiplos abscessos est?o presentes e quando as crian?as s?o menores de 1 ano.
Epilepsia pode desenvolver após o abscesso.
A convuls?o febril (C.F.) é um evento próprio da infancia, geralmente ocorrendo entre 3 meses e 5 anos de idade, associada à febre, mas sem evidência de infec??o intracraniana ou causa definida.
S?o excluídas da defini??o aquelas crian?as que tiveram convuls?es afebris previamente.
O baixo limiar da córtex em desenvolvimento, a suscetibilidade da crian?a a infec??es, a propens?o alta e o componente genético afetando o limiar convulsígeno s?o fatores que combinam e justificam porque a convuls?o febril é um fen?meno da primeira infancia e é sobrepujado com o crescimento.
A- Simples: se a crise for generalizada, com dura??o inferior a 15 minutos, n?o recorrer em 24 horas e n?o apresentar anormalidades neurológicas pós-ictal.
B- Complexa ou complicada: se a crise for focal ou durar mais do que 15 minutos ou recorrer em 24 horas.
Um estudo epidemiológico feito no Chile revelou uma incidência de Convuls?o Febril na popula??o infantil de 4%.
Esta taxa, provavelmente se aproxime da nossa realidade.
O prognóstico a longo prazo é favorável. Há história de óbito, seqüelas motoras, ou prejuízo intelectual.
Os déficits cognitivos s?o detectados apenas nas crian?as que já apresentavam comprometimento neurológico prévio à convuls?o febril.
Os prováveis problemas que podem acometer as crian?as que tiveram uma convuls?o febril s?o: recorrência da crise e/ou posterior epilepsia.
Aproximadamente, um ter?o das crian?as que tiveram uma C.F. ter?o uma ou mais C.F. recorrentes.
Crian?as com até 12 meses, a taxa de recorrência passa para 50%; se a faixa etária for até 18 meses, o risco de recorrência passa para 40%.
Os fatores preditivos para a recorrência s?o:
a- idade da primeira crise inferior a 15 meses;
b- epilepsia em parentes de primeiro grau;
c- C.F. em parentes de primeiro grau;
d- primeira C.F. do tipo complexa.
O risco de epilepsia que se segue à C.F. é bastante baixo e variável.
Os fatores de risco para a epilepsia s?o:
a- história familiar de epilepsia;
b- anormalidade neurológica;
c- C.F. complexa;
d- número aumentado de recorrência.
Existe estreita correla??o entre C.F. e esclerose mesial hipocampal e discute-se, na literatura, o que é causa ou conseqüência.
A ocorrência de estado de mal febril em uma crian?a normal, n?o aumenta significantemente o risco para subseqüentes C.Fs. ou crises afebris nos primeiros anos que se seguem ao evento.
O melhor tratamento para crian?as que tiveram a primeira C.F. n?o é a prescri??o medicamentosa, mas sim, a conversa com os pais, procurando informá-los e acalmá-los, assegurando-lhes que a grande maioria das C.Fs. s?o únicas, n?o causando dano físico e n?o necessitam de tratamento.
N?o há qualquer evidência de que o tratamento prolongado com anticonvulsivante previna o desenvolvimento de posterior epilepsia.
Indica??o de tratamento:
a- idade precoce da C.F. (< 18 m);
b- história familiar positiva para C.F.;
c- febre baixa com dura??o inferior a uma hora antes da crise.
Tratamento:
a- Profilaxia contínua:
A.1- Fenobarbital- tem efeitos colaterais, como: hiperatividade, instabilidade emocional, agressividade, distúrbio do sono, sonolência excessiva, eritema cutaneo e diminui??o da performance cognitiva.
- Dose = 5mg/Kg/dia de 12/12 horas ou 1 vez por dia.
A 2- Valproato- tem efeitos colaterais, como: sintomas gatrointestinais, seda??o, ataxia, eritema cutaneo e hepatite fulminante.
- Dose = 10 –20mg/Kq/dia de 8/8 horas.
b- Profilaxia Intermitente:
B 1- Diazepam oral- dado somente quando a febre está presente.
- Dose = 0,5-1mg/ Kg/ dia de 12/12 horas.
- O efeito colateral é leve ou moderado, como: agita??o, sonolência ou ataxia, que desaparecem com a suspens?o do tratamento.
O objetivo aqui é mostrar ao leigo e ao clínico de outra especialidade uma vis?o ampla e clara do assunto.
? O que é epilepsia?
? O que causa a epilepsia?
? Como reconhecer uma crise epiléptica?
? O que fazer durante uma crise epiléptica?
? Como é o tratamento?
? A crian?a com epilepsia pode praticar esportes?
? Qual a conduta a ser tomada nas escolas?
? Qual é a finalidade do eletroencefalograma?
? Conclus?o
A epilepsia é um distúrbio do cérebro e n?o uma doen?a mental.
As crises epilépticas s?o eventos clínicos, que refletem uma disfun??o temporária de uma pequena área do cérebro (crises focais), ou de área mais extensa, envolvendo os dois hemisférios cerebrais ( crises generalizadas), causada por uma descarga anormal excessiva e transitória das células nervosas.
As crises epilépticas podem ser desencadeadas por varias causas, como: febre alta, infec??o do Sistema Nervoso Central, intoxica??o, trauma craniano e outros.
Os sintomas dependem das partes do cérebro envolvidas na disfun??o.
O primeiro passo no diagnóstico é definir se os episódios s?o epilépticos ( repetitivos ) e ent?o, tentar identificar a causa.
O médico deve obter a maior quantidade possível de dados, pois o diagnóstico de epilepsia é basicamente, clínico - presuntivo.
A história deve incluir a descri??o dos sintomas que precedem a crise epiléptica ( ocorrência de aura ou aviso), as manifesta??es durante a crise ( área do corpo inicialmente, afetada; progress?o da atividade e sua evolu??o e o momento de ocorrência no dia) e os sintomas e sinais após a crise.
Devem ser observados:
? movimentos involuntários, como estalar os lábios, mastiga??o ou careta;
? movimentos involuntários dos membros, sem conseguir parar;
? movimentos oculares - revira os olhos e pisca com muita freqüência;
? altera??o da consciência;
? libera??o esfincteriana ( urinar - evacuar );
? apnéia ( parar de respirar );
? cianose;
? quedas súbitas sem raz?o aparente;
? mudan?a de comportamento - age de forma estranha e n?o natural;
? mordedura de língua;
? distúrbio de linguagem;
? dores de cabe?a;
? quedas súbitas da cabe?a momentaneamente e , depois, volta ao normal;
? pára, momentaneamente suas atividades e fica com o olhar perdido.
O exame neurológico convencional é, geralmente normal em pacientes com epilepsia. A presen?a de anormalidades sugere, que as crises s?o secundárias à doen?a cerebral organica
? Mantenha o paciente calmo;
? coloque algo suave sob a sua cabe?a;
? vire, suavemente a cabe?a para o lado, de maneira que a saliva escorra e n?o impe?a a respira??o;
? n?o coloque nenhum objeto na boca;
? n?o jogue água nele;
? permane?a ao lado do paciente até terminar a crise.
Após a avalia??o médica, constatando-se a epilepsia será receitado o medicamento específico.
N?o suspenda a medica??o e nem aumente a dose por conta própria, sem falar com o seu médico.
Deve ser considerado o risco, de acordo com o tipo de epilepsia.
Se as crises est?o controladas, é possível a prática de esportes sob supervis?o.
é importante, que a crian?a aprenda a conviver com a epilepsia, tratando esse assunto de forma natural.
A crian?a deve se sentir igual aos outros colegas, tendo os mesmos direitos e respeitando as mesmas regras escolares.
Solicite aos professores, observa??es sobre a aten??o, dificuldade de aprendizagem, crises em sala de aula e o seu relacionamento com outras crian?as, para que essas dificuldades sejam trabalhadas adequadamente.
E.E.G. ainda é o exame de maior sensibilidade na avalia??o das epilepsias.
Tem a finalidade de:
? Confirmar o diagnóstico clínico;
? Ajudar na classifica??o das crises e das síndromes epilépticas;
? Fornecer informa??o prognóstica.
Com o diagnóstico e tratamento adequados, aproximadamente 80% - 90% dos pacientes ter?o suas crises controladas, com um mínimo de efeitos indesejados, dando-lhes uma vida normal.
A Organiza??o Mundial de Saúde define epilepsia como uma desordem cerebral cr?nica, de várias etiologias, caracterizada por crises recorrentes conseqüentes a descargas neuronais excessivas (crises epilépticas).
As crises parciais apresentam diferentes express?es clínicas, dependendo da área cerebral envolvida e s?o caracterizadas no EEG por descargas focais.
Uma síndrome é considerada benigna, se a crian?a apresentar os exames físico e neuropsicológicos normais, outros exames, com exce??o do EEG, normais, boa resposta a terapia anticonvulsivante, baixa freqüência das crises e, o curso clínico tender para a remiss?o completa, sem riscos de deteriora??o neuropsíquica. Portanto, é necessário considerar os dados clínicos e eletrencefalográficos do início do processo, e observar atentamente sua evolu??o.
Logo, a combina??o de uma crian?a normal, com crises raras e atividade epileptiforme focal, desproporcionalmente freqüente, é muito sugestiva de epilepsia parcial benigna da infancia.
A Epilepsia Parcial Benigna da Infancia com paroxismos centro-temporais é a forma mais comum de epilepsia na infancia, correspondendo a 75% das epilepsias benignas da infancia.
? idade de início - 3-13 anos (pico - 9-10 anos) e recupera??o até 15-16 anos;
? predisposi??o genética - é controlado por um único gene autoss?mico dominante, penetrancia idade dependente;
? os meninos s?o mais afetados que as meninas - (60% homens, 40% mulheres);
? s?o crian?as previamente normais, sem evidência de les?o cerebral e apresentam uma história familiar rica (convuls?o febril; crises parcias ou generalizadas; descargas epileptiformes focais ou generalizadas no EEG, sem crises clínicas);
? início somatossensorial com parestesias unilaterais envolvendo a língua, lábios, gengiva e bochechas, associadas a crises motoras hemifaciais, breves, simples, com convuls?o t?nico - cl?nica, t?nica ou cl?nica, envolvendo a face, lábios, língua, músculo da faringe e laringe, que causam anartria e hipersaliva??o; a crise pode estender ao bra?o (crise bráquio-facial) e raramente a perna (crise unilateral);
? consciência preservada.
? o episódio n?o dura mais que 1 a 2 minutos. Crises longas s?o raras e podem ser seguidas de paralisia de Todd;
? crise noturna é a mais freqüente e pode se tornar generalizada (15 a 20%);
? a express?o das crises parecem ser idade dependente (crian?as mais velhas- crises puras hemifaciais s?o mais comuns, enquanto crian?as mais novas, podem apresentar mais chance de hemiconvuls?o e convuls?o noturna generalizada);
? variam em freqüência. Em 20% dos casos, as crises podem ser freqüentes e de difícil manejo, mesmo após a institui??o da medica??o correta, mas isso n?o influencia o prognóstico excelente desta síndrome;
respondem muito bem as drogas antiepilépticas. Geralmente, é introduzida a Carbamazepina.
Anormalidades típicas no EEG de superfície s?o essenciais para a confirma??o do diagnóstico.
Características:
O tra?ado eletrencefalográfico é caracterizado por uma atividade elétrica cerebral de base normal. Em alguns casos, a ocorrência quase contínua das descargas pode mostrar um pseudo -alentecimento, tendo que se fazer o diagnóstico diferencial de les?o estrutural.
Durante o registro, observam -se descargas paroxísticas de pontas (<70ms) ou ondas agudas (<200ms) de amplitude variável (50 -300 Uv ou mais), rombas e difásicas, ora seguidas por onda lenta proeminente. Podem ser unilaterais (60%; ocorrem em igual propor??o no hemisfério direito e esquerdo, contra ou ipsilateral às manifesta??es clínicas), bilaterais (40%; podem ser síncronas ou assíncronas), ou multifocais (37%; devem ser diferenciadas de outras formas de epilepsias).
Normalmente, s?o localizadas em regi?es centrais ou mediotemporais, recorrendo em intervalos curtos, freqüentemente em grupos, de 2-3 elementos, por vários segundos. S?o ativados pela sonolência e pelo sono (todos os estágios, inclusive sono Rem), quando tendem a se tornar bilaterais. Em 30% dos casos, as descargas s?o exclusivamente durante o sono. Portanto, EEG em sono é fundamental para a caracteriza??o desta síndrome. A abertura ocular, a fotoestimula??o e a hiperventila??o n?o provocam nenhum efeito nestas descargas.
Freqüentemente, s?o vistos dipolos tangenciais à superfície cortical (pólo negativo nos eletrodos centro - temporais e um pólo positivo no eletrodo frontal superior).
O pólo negativo, tende a difundir - se às regi?es têmporo - parieto - auricular, enquanto o pólo positivo para as áreas fronto -polares. A identifica??o destes dipolos é melhor realizada através do EEG digital e do Mapeamento Cerebral, do que pelo EEG convencional, que é caracterizado pela presen?a do padr?o n?o - estacionário ou dupla ponta - onda, onde faz - se o diagnóstico diferenciada epilepsia parcial secundária. A morfologia característica das pontas e a topografia dipolar de seu campo elétrico s?o mais associadas com epilepsia benigna do que sintomática.
Legarda e col. Estudaram 33 crian?as com epilepsia rolandica, cujos EEG em montagens com o sistema 10-10 mostraram que em 10 crian?as, predominava a negatividade em C3-C4 e, em 23 crian?as em C5-C6 e, nenhuma em T3-T4.
Em 10-20% dos casos, as descargas s?o encontradas em outras localiza??es. As descargas focais podem se apresentar como focos únicos máximos na regi?o centrotemporal, temporoparietal ou parietal. Há varia??o na localiza??o, em EEGs obtidos em diferentes épocas. O conceito de epilepsia rolandica n?o é ligado a uma localiza??o específica das descargas, mas a sua morfologia típica ou sugestiva.
A normaliza??o do EEG ocorre em 6 meses a 6 anos; primeiro, do tra?ado de vigília e só depois durante o sono, após a remiss?o clínica (em média em 2 anos). Ent?o, a persistência de anormalidades no EEG n?o necessariamente, contra-indica a descontinuidade do tratamento.
Pode-se encontrar descargas semelhantes em crian?as sem nenhuma história clínica de crises. é reconhecido, que a freqüência das crises n?o tem nenhuma rela??o com a freqüência das descargas.
1.WORD HEALTH ORGANIZATION. Dictionary of Epilepsy. 1973. WHO, Geneva.
2.YACUBIAN, E.M.T.- Síndromes Epilépticas - Aspectos Clínicos. BJECN 3 (1) 33 - 44, 1997.
3.SILVA, D.F. e col. - Mapeamento Cerebral. Ver. Neurociências 3 (1) 11-18, 1995.
4.SAKAMOTO, A . C. E col -EEG nas Epilepsias e Síndromes Epilépticas. In: Guerreiro C. A .M. e Guerreiro M.M. Epilepsia 2a Edi??o. Lemos, S?o Paulo 1996: 41-67.
5.SILVA, E.S.M. e col - Epilepsia Benigna da Infancia. In: Guerreiro C. A .M. e guerreiro M.M. Epilepsia 2 Edi??o. Lemos, S?o Paulo 1996: 133-151.
6.MEIJ, W.V. e col - Sequential EEG mapping may diferentiate “epileptic” from “non-epileptic” rolandic spikes. Eletrencephalography and clinical Neurophysiology, 82 (1992) 408-411,1992.v
7.D.L.Gregory e col - Clinical Relevance of a Dipole Field in Roland Spikes. Epilepsia, 33 (1): 36-44,1992.
8.D.L.Gregory e col - Topographical Analysis of the Centrotemporal Discharges in Benign Rolandic Epilepsy of Chilhood. Epilepsia, 25 (6): 705-711,1984.
9.LEGARDA, S. e col - Benign Roland Epilepsy: High Central and Low Central Subgroups. Epilepsia 1994; 35 (6): 1125-1129.
Garrod, há quase 100 anos, introduziu o conceito de erros inatos do metabolismo e de individualidade química, onde identificou mais de 300 defeitos genéticos relacionados a síntese, metabolismo, transporte e armazenamento dos compostos bioquímicos.
As perturba??es metabólicas agudas s?o eventos relativamente freqüentes na infancia, e suas causas mais comuns s?o as infec??es, a imaturidade e as disfun??es ventilatórias e hidroeletrolíticas. Porém, num pequeno grupo de pacientes, elas s?o decorrentes de uma doen?a metobólica primária - os Erros Inatos do Metabolismo.
A express?o distúrbio neuro-metabólico é aplicada a mais de 100 erros do metabolismo, associados à anormalidades neurológicas. A freqüência deste distúrbio metabólico no Sistema Nervoso Central é estimada em mais de 30%.
As manifesta??es dos Erros Inatos do Metabolismo de instala??o aguda s?o extremamente inespecíficas, tornando o seu diagnóstico uma tarefa de interpreta??o laboratorial.
A demora no diagnóstico e tratamento dos E.I.M. pode trazer danos irreparáveis ao Sistema Nervoso Central. Por outro lado, a detec??o e a interven??o precoces levam a uma evolu??o clínica favorável e a preven??o de novos afetados.
As tentativas de classificar os E.I.M., com comprometimento neurológico, de acordo com os achados clínicos, incluem:
a - Localiza??o anat?mica (substancias cinzenta e branca ):
? Aquelas que afetam principalmente a substancia cinzenta apresentam-se com anormalidades de cogni??o ( retardo mental ), da vis?o, da audi??o, e convuls?es;
? As que afetam principalmente a substancia branca apresentam-se com perda da capacidade motora, espasticidade ou ataxia.
b - Intoxica??es e deficiência energética:
? Intoxica??es - resultam do acúmulo de compostos tóxicos anteriores ao bloqueio metabólico - acidúrias organicas, aminoacidopatias, defeitos do ciclo da uréia, galactosemia, frutosemia e tirosinemia.
? Deficiência energética - é devido a produ??o ou utiliza??o de energia diminuídas, em virtude de defeitos no fígado, miocárdio, músculo ou cérebro - distúrbio do armazenamento do glicogênio, acidose lática congênita, defeito da oxida??o dos ácidos graxos, distúrbios da respira??o mitocondrial e os distúrbios peroxiss?micos.
c - Doen?as das pequenas e grandes moléculas:
? Pequenas moléculas - caracterizam-se por episódios agudos no período perinatal ou nos primeiros meses de vida - aminoácidos, ácidos organicos e a?úcares.
? Grandes Moléculas - tendem a ocorrer nos lactentes mais velhos e crian?as, sob a forma de processos cr?nicos degenerativos progressivos - glicogênios, lipídios e mucopolissacarídios.
d- Idade de aparecimento - neonatal;
1-12 meses;
1-4anos;
5-15 anos.
e- Manifesta??es neurológicas;
? Encefalopatias agudas- ocorrem no início da vida. Evoluem com v?mitos, letargia, má alimenta??o e desidrata??o. S?o rapidamente progressivas, podendo desenvolver altera??o do t?nus, convuls?o, altera??o respiratória, coma e, eventualmente a morte. Causadas por doen?a de pequenas moléculas, afeta principalmente substancia cinzenta e representam um quadro de intoxica??o;
? Encefalopatias cr?nicas ou progressivas - est?o presentes no lactente, na infancia ou na adolescência. Seu aparecimento é gradual, com espasticidade ou ataxia e demência progressiva. O fígado, o cora??o, o músculo ou os rins podem estar envolvidos. S?o decorrentes de altera??es de grandes moléculas, substancia branca e intoxica??o.
? Aminoacidopatias- 31,3%
? Acidúrias organicas- 27%
? Hiperamonemias -20,8%
? Cadeia respiratória- 12%
? Distúrbio do armazenamento do Glicogênio -8,3%
? Oxida??o de ácidos graxos- 8,3%
? Distúrbios peroxiss?micos- 4,6%
As manifesta??es clínicas dos EIM costumam ser divididas, de um modo geral, em 4 grandes grupos:
1) o dos sintomas agudos no período neonatal;
2) o dos sintomas agudos e freqüentemente intermitentes, de apresenta??o tardia (> 29 dias );
3) o dos quadros específicos que envolvem deteriora??o progressiva de determinado órg?o ou sistema;
4) o dos quadros cr?nicos gerais e inespecíficos .
Os seguintes critérios de identifica??o de crian?as com risco de serem portadoras de um E.I.M. de manifesta??o aguda s?o:
1- Sintomas iniciais de recusa alimentar, v?mitos, letargia,e às vezes hipotonia marcada. Isto traduz, o que corresponderia à irritabilidade das fases iniciais de uma Encefalopatia Aguda e à avers?o a algum componente da dieta, que fosse precursor de substancia tóxica. A progress?o destes sintomas costuma evoluir para o coma, para crises convulsivas e para perturba??es ventilatórias, desde taquipnéia, por acidose metabólica ou por estímulo direto dos centros ventilatórios, até parada respiratória.
2- Crian?as que, em associa??o com isso, apresentem odores peculiares, cataratas, visceromegalias, ou dismorfias.
3- Crian?as nas quais se identifiquem uma modifica??o alimentar ou uma situa??o de catabolismo subjacente.
4- História de recorrência familiar ou de consangüinidade entre os pais.
Os exames laboratoriais permitem um rastreamento de várias doen?as ou de um grupo específico de E.I.M.
A conduta inicial é a investiga??o bioquímica, na suspeita de uma doen?a neuro-metobólica, que só será eficaz se as amostras de sangue e urina forem colhidas antes de qualquer manejo terapêutico.
As primeiras condutas clínicas frente a um bebê com v?mitos, apnéia, coma, etc. sempre incluem:
1- suspens?o da via oral;
2- administra??o de solu??es parenterais (glicose, eletrólitos e às vezes bicarbonato, se houver acidose metabólica).
Tempo zero- na chegada ao hospital ou no início do quadro:
a) Urina - corpos cet?nicos (quantificar);
- substancias redutoras;
- cetoácidos;
- pH;
- cromatografia de aminoácidos;
- amostra para congelamento imediato (-20O.C, maior volume possível);
b) Sangue - gasometria arterial (principalmente para pCO2, HCO3 e pH);
- Sódio e cloretos para cálculo de anion gap (> 16 nas acidemias organicas), glicose e cálcio;
- am?nia;
- lactato;
- cromatografia de aminoácidos;
- amostras para congelamento imediata (separar 5 ml de plasma, n?o congelar o sangue total).
Tempo um - tratamento agudo (após a coleta e antes de seus resultados):
- interrup??o da ingesta proteica. Após alguns dias, iniciar com 0,5 a 1 g/kg/dia de proteina;
- oferta aumentada de energia (indi??o de anabolismo com administra??o de glicídio E.V. ou V.O, glicoinsulinoterapia, uso de GH).
- corre??o da acidose metabólica, se houver, com administra??o de bicarbonato;
- controle da hiperamonemia através da diálise peritoneal ou exangüíneo-transfus?o, se am?nia >400mmoles/L. Cuidado com o edema cerebral secundário a hiperamonemia;
- uso de cofatores:
? 1- Piridoxina VO ou EV, 10 a 100mg/dia, para bebês com convuls?es;
? 2- coquetel de cofatores deve ser iniciado, com cada unidade contendo o seguinte: a. fórmula EV (conteúdo de 1 unidade- para crian?as de 3 a 6 Kg)- Vitamina B1- 100 mg, Biotina- 10 mg, Vitamina B2- 50 mg, L- carnitina- 100 mg/kg/dia;
b. fórmula IM- Vitamina B12- 1 mg/dia IM.
O resultado obtido nos exames laboratoriais permite a classifica??o do distúrbio neuro-metabólico em um dos cincos grupos possíveis:
a - Doen?as neurometabólicas associadas à cetose:
? é representada pela doen?a da urina em xarope de bordo;
? Achados clínicos - má alimenta??o;
? v?mitos;
? choro em tom agudo;
? altera??o entre hipertonia e hipotonia;
? convuls?o;
? perda dos reflexos tendíneos profundos;
? coma;
? acidose metabólica e hipoglicemia grave;
? odor característico na urina de cury ou de xarope de bordo;
? Diagnóstico - análise quantitativa de aminoácidos plasmáticos, dos ácidos organicos na urina e de enzimas específicas nos fibroblastos da pele.
b - Doen?a neurometabólica com cetoacidose e acidose;
? Este grupo inclui a maioria das acidúria organicas (acidúria metilmal?nica, propi?nica e isovalérica) e a deficiência múltipla de carboxilase.
? Distúrbio V?mitos Má Problemas Encefalopatia Odor alimenta??o respiratórios urinário
? Acidúria Metilmal?nica + + + + +
? Acidúria propi?nica + + + + -
? Acidúria Isovalérica + + + + +
? Dficiência múltipla + + + + - de carboxilase
? Diagnóstico: efetuado através dos ácidos organicos na urina e das enzimas específicas em fibroblastos cultivados.
c - Doen?as neurometabólicas com acidose lática;
? Distúrbios Encefalopatia V?mitos Anormalidades Insuficiência Miopatia
? Recorrentes Respiratórias Hepática
? Doen?a de Leigh + - + - -
? Doen?a de Alpert + + - + -
? Sínd. De Kearns-Sayre + - - - +
? MERFF + - - - +
? MELAS + + - - +
? Diagnóstico: análise enzimática utilizando tecido muscular ou fibroblasto cultivado.
d - Doen?as neurometabólicas com Hiperamonemia e sem Cetoacidose
? As altera??es no ciclo da uréia nas crian?as, geralmente aparecem entre os dois à quatro anos de idade, em forma de um transtorno metabólico grave.
? A hiperamonemia sintomática é uma emergência médica que deve ser reconhecida de maneira precoce, diagnosticada de maneira específica, e tratada de forma agressiva.
? Os primeiros sintomas consistem em dificuldade na alimenta??o, hipotonia, v?mitos, letargia, altera??o respiratória e convuls?o. Esta sintomatologia progride, rapidamente ao coma e a morte.
? A apresenta??o clínica se associa com níveis de am?nia plasmática entre 500-2000umol/l.
? é necessário instituir, de maneira imediata, algumas medidas terapêuticas.
e - Doen?as neurometabólicas sem cetoacidose e sem hiperamonemia.
? Os distúbios mais encontrados s?o: hiperglicinemia n?o-cetótica, deficiência da sulfato- oxidase e os distúbios peroxiss?micos.
? Diagnóstico: exame dos sulfatos urinários (deficiência de sulfato oxidase), dos níveis cerebroespinais de glicina (hiperglicinemia n?o-cetótica) e dos ácidos graxos de cadeia muito longa (distúrbios peroxiss?micos).
Com rotinas estabelecidas, em especial no que se refere ao primeiro momento, logo após a chegada do paciente ao hospital (tempos zero e um), acreditamos que um número cada vez maior de crian?as gravemente enfermas, receberá diagnósticos mais precisos e tratamentos mais eficazes.
Nosralla Medicina e Odontologia - Av. Brig. Luiz Antônio, 3333 - cjs. 51 e 52 - Jardim Paulista - São Paulo - SP.


